assoz. Prof. Dr. Thomas Hofer
Fortgeschrittene Quantenchemie und theoretische Materialwissenschaften

Computergestützte Methoden sind unverzichtbare Werkzeuge in der modernen Chemie und Materialforschung, da sie eine Reihe von Vorteilen im Forschungalltag bieten. Die computergestützte Chemie ermöglicht die Simulation komplexer chemischer Prozesse und des Verhaltens molekularer Systeme, die experimentell oft nur schwer oder gar nicht zu untersuchen sind. Quantenchemische Methoden liefern darüber hinaus detaillierte Informationen über die elektronische Struktur und die Eigenschaften von Molekülen und Materialien auf atomarer Ebene. Mit Hilfe dieser Verfahren lässt sich eine große Anzahl chemischer Verbindungen und Materialien effektiv und zeitsparend untersuchen, wodurch in vielen Fällen teure und zeitaufwändige experimentelle Versuche vermieden werden können. Die vielversprechendsten Kandidaten können dann von den experimentellen Arbeitsgruppen im Labor weiter untersucht werden.

Die Entwicklung neuartiger, computergestützter Methoden in den Materialwissenschaften ist ein aktives Forschungsthema der Arbeitsgruppe. Schlüsseltechniken wie die Dichtefunktionaltheorie (DFT) und die Molekulardynamik (MD) bilden die Grundlage für diesen Bereich. DFT ermöglicht die quantenmechanische Modellierung elektronischer Eigenschaften, während MD-Simulationen Einblicke in die physikalischen Bewegungen von Atomen und Molekülen liefern. Die Kombination dieser beiden Methoden ermöglicht detaillierte Computersimulationen von Molekülen und Festkörpern, die Informationen über strukturelle, dynamische und thermodynamische Eigenschaften liefern

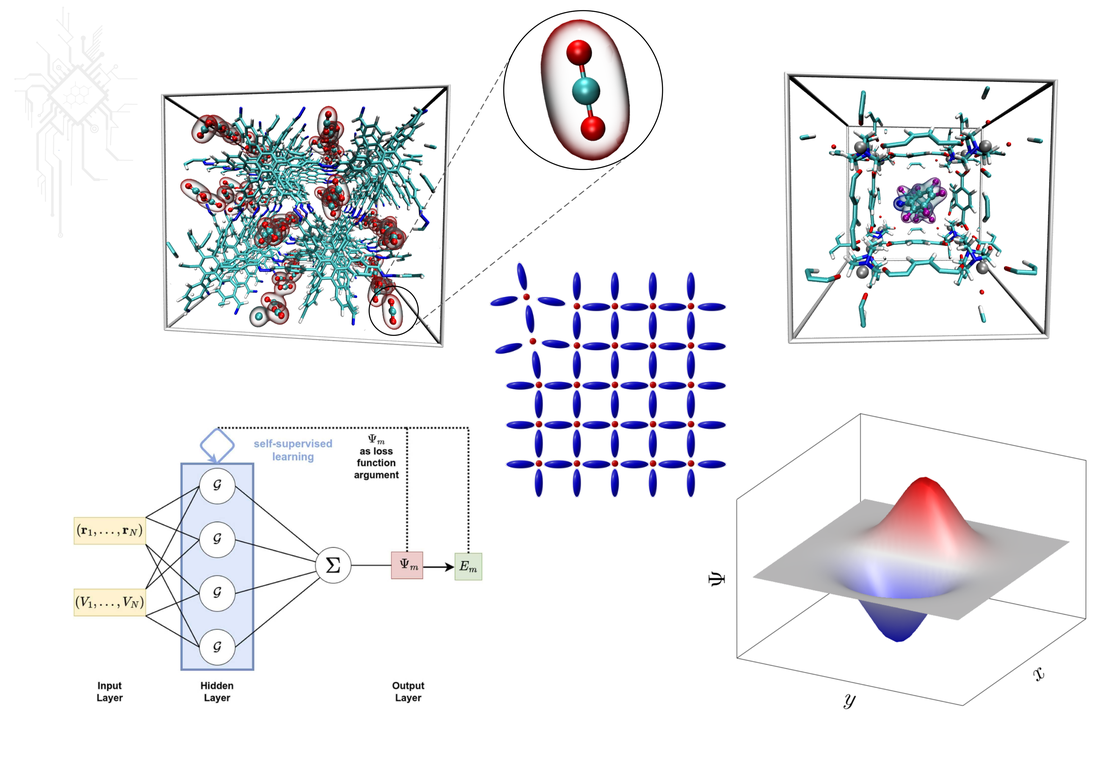
Speicherung des klimaschädigenden Gases Kohlendioxid (CO2) in der metall-organischen Gerüstverbindung MOF-5. Die Interaktion zwischen den CO2 Gastmolekülen und dem Speichermaterial kann effizient und genau mit Hilfe computergestützter Methoden modelliert werden.
Metallorganische Gerüsteverbindungen und andere Funktionsmaterialien haben einzigartige physikalische und chemische Eigenschaften, die ihnen besondere Fähigkeiten verleihen. Diese Materialen kombinieren die geordnete Struktur eines Kristalls mit der Flexibilität und Bewegung einer Flüssigkeit. Sie können molekulare Schalter und Maschinen enthalten, die es dem Material ermöglichen, auf äußere Reize zu reagieren. Das Verständnis der Beziehungen zwischen der Struktur, der Dynamik und der Thermodynamik dieser Materialien ist der Schlüssel zur Entwicklung neuer funktioneller Materialien für Anwendungen. Die Erforschung dieser komplexen Verbindungen erfordert einen interdisziplinären Ansatz, der Chemie, Physik, Materialwissenschaft und Computermodellierung miteinander verbindet. Mit den Fortschritten auf diesem Gebiet werden neue funktionelle Materialien mit innovativen Eigenschaften und Verwendungen möglich.

Aufbau eines physik-informierten neuronalen Netzwerks (PINN) zur Lösung der Schrödingergleichung (oben). Beschreibung eines quantenmechanischen Tunneleffekts am Beispiel der OH-Torsionsmode im Phenolmolekül (unten).
Maschinelles Lernen (ML) ist ein modernes und leistungsfähiges Werkzeug, das die Quantenchemie und die Materialwissenschaften revolutioniert. Durch die Entwicklung von Algorithmen auf Grundlage großer Datenmengen können Materialeigenschaften effizient und mit hoher Geschwindigkeit vorhergesagt werden. Mit den Modellen des maschinellen Lernens kann beispielsweise die Energie von Molekülen errechnet werden, was wesentlich zum Verständnis chemischer Reaktionen beiträgt. Die Integration des maschinellen Lernens mit traditionellen Berechnungsmethoden wie der Quantenmechanik ermöglicht einen synergetischen Ansatz, der die Stärken beider Techniken kombiniert. Da ML-basierte Algorithmen ständig verbessert werden, ist bereits in der nahen Zukunft mit Durchbrüchen beim Design und der Entwicklung neuer funktionaler Materialien zu rechnen.