assoz. Prof. Dr. Thomas Hofer
Advanced Quantum Chemistry and Computational Material Sciences

Computational methods are indispensable tools in modern chemistry and materials research, as they offer a number of advantages in day-to-day research. Computational chemistry makes it possible to simulate complex chemical processes and the behaviour of molecular systems that are often difficult or impossible to investigate experimentally. Quantum chemical methods also provide detailed information about the electronic structure and properties of molecules and materials at the atomic level. With the help of these methods, a large number of chemical compounds and materials can be evaluated effectively and in a time-saving manner, which in many cases means that expensive and time-consuming experimental tests can be avoided. The most promising candidates can then be further investigated by the experimental working groups in the laboratory.

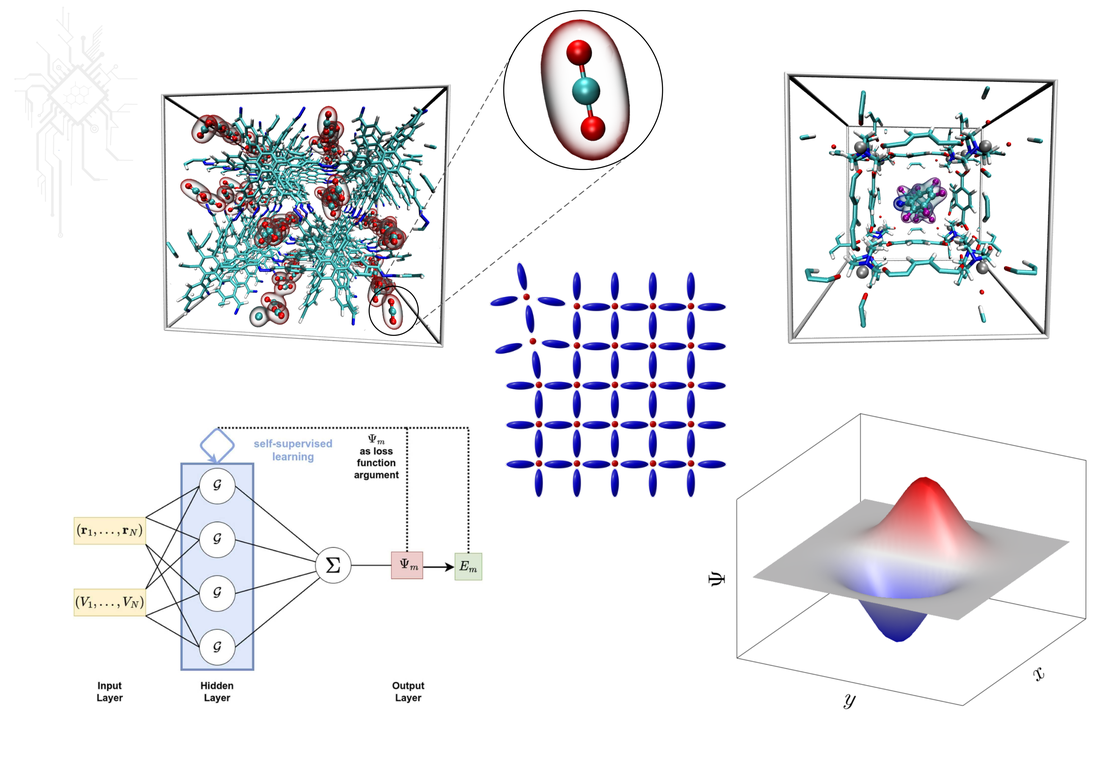
The development of novel, computer-aided methods in materials science is an active topic of the research group. Key techniques such as density functional theory (DFT) and molecular dynamics (MD) form the basis for this field. DFT enables quantum mechanical modelling of electronic properties, while MD simulations provide insights into the physical movements of atoms and molecules. The combination of these two methods enables detailed computer simulations of molecules and solids, which provide information on structural, dynamic and thermodynamic properties.

Storage of the climate-damaging gas carbon dioxide (CO2) in the metal-organic framework compound MOF-5. The interaction between the CO2 guest molecules and the storage material can be modelled efficiently and accurately using computer-aided methods.
Metal-organic frameworks (MOFs) and other functional materials have unique physical and chemical properties that give them special capabilities. MOFs combine the ordered structure of a crystal with the flexibility and motion of a liquid. They can incorporate molecular switches and machines that allow the material to respond to external stimuli. Understanding the relationships between the structure, dynamics, and thermodynamics of these compounds is key to designing new functional materials for applications. Studying these complex structures requires an interdisciplinary approach combining chemistry, physics, materials science and computational modeling. As the field advances, it will enable new functional materials with innovative properties and uses.

Structure of a physics-informed neural network (PINN) for solving the Schrödinger equation (top). Description of a quantum mechanical tunnelling effect at the example of the OH torsional mode in the phenol molecule (bottom).
Machine learning (ML) is a modern and powerful tool that is revolutionising quantum chemistry and materials science. By developing algorithms based on large amounts of data, material properties can be predicted efficiently and at high speed. For example, machine learning models can be used to calculate the energy of molecules, which contributes significantly to the understanding of chemical reactions. The integration of machine learning with traditional computational methods such as quantum mechanics enables a synergistic approach that combines the strengths of both techniques. As ML-based algorithms are constantly being improved, breakthroughs in the design and development of new functional materials can be expected already in the near future.